
基因外显子组测序技术手册
2014-02-07 千年基因 千年基因

外显子组的序列仅占全基因组序列的1%左右,但大多数与疾病相关的变异位于外显子区。通过外显子组测序可鉴定约8万个变异,全基因组测序可鉴定300 万个变异,因此,与全基因组测序相比,外显子组测序不仅费用较低,数据阐释也更为简单。外显子组测序技术以其经济、有效的优势广泛应用于孟德尔遗传病、罕 见综合征及复杂疾病的研究,并于2010年被Science杂志评为十大突破之一。 一、技术简介 随
外显子组的序列仅占全基因组序列的1%左右,但大多数与疾病相关的变异位于外显子区。通过外显子组测序可鉴定约8万个变异,全基因组测序可鉴定300
万个变异,因此,与全基因组测序相比,外显子组测序不仅费用较低,数据阐释也更为简单。外显子组测序技术以其经济、有效的优势广泛应用于孟德尔遗传病、罕
见综合征及复杂疾病的研究,并于2010年被Science杂志评为十大突破之一。
一、技术简介
随着社会生活水平的提高,人类健康问题也越来越多的受到社会各界的关注。传统的遗传疾病研究模式是采用显带 分析、核型分析、FISH、遗传标记、PCR-DNA测序等传统试验方法来寻找与疾病相关的DNA变异,这些方法各有各的特点,但都存在工作量大、效率 低、分辨率低等一系列的限制。新一代高通量测序技术的出现,为遗传疾病的研究提供了全新的思路。
2009年,基因组定向捕获工具的出现使外显子组测序成 为可能。2009年9月,第一篇关于外显子组测序的原理验证文章于Nature杂志上发表。来自华盛顿大学的Jay Shendure通过对四名Freeman-Sheldon综合征患者的外显子组测序,找到了已知的致病基因MYH3。随后,该团队将这种技术应用于米勒 综合征的研究,通过对患者编码区序列的捕获及深度测序,鉴定出单个候选基因DHODH,并经Sanger测序验证其他患者中存在该基因的突变。
外显子组的序列仅占全基因组序列的1%左右,但大多数与疾病相关的变异位于外显子区。通过外显子组测序可鉴 定约8万个变异,全基因组测序可鉴定300万个变异,因此,与全基因组测序相比,外显子组测序不仅费用较低,数据阐释也更为简单。外显子组测序技术以其经 济、有效的优势广泛应用于孟德尔遗传病、罕见综合征及复杂疾病的研究,并于2010年被Science杂志评为十大突破之一。近两年外显子组研究相关的 SCI文章已发表千余篇,已对数百种疾病展开了深入研究,研究结果推动了人类医学的研究。
二、技术优势
• 直接对蛋白编码序列进行序列测定,找出影响蛋白结构的变异。
• 高深度测序,可发现常见变异及频率低于1%的罕见变异。
• 针对外显子组区域测序,约占基因组的1%,有效降低费用、周期、工作量。
三、应用举例
|
疾病 |
遗传模式 |
致病基因 |
|
Freeman-Sheldon综合征 |
AD |
MYH3 |
|
Kabuki 综合征 |
AD |
MLL2 |
|
Schinzel-Giedion 综合征 |
AR |
SETBP1 |
|
Sensenbrenner 综合征 |
AR |
WDR35 |
|
Fowler 综合征 |
AR |
FLVCR2 |
|
Perrault 综合征 |
AR |
HSD17B4 |
|
Hajdu-Cheney 综合征 |
AD |
NOTCH2 |
|
成骨不全 |
AR |
SERPINF1 |
|
米勒综合征 |
AR |
DHODH |
|
Brown-Vialetto-van Laere 综合征 |
AR |
C20orf54 |
|
血磷酸脂酶过多智力迟钝综合征 |
AR |
PIGV |
|
家族性β-脂蛋白过少血症 |
AD |
ANGPTL3 |
|
色素性视网膜炎 |
AR |
DHDDS |
|
非综合征性耳聋 |
AR |
GPSM2 |
|
原发性淋巴管性水肿 |
AD |
GJC2 |
|
肌萎缩性侧索硬化 |
AD |
VCP |
|
非综合征的智力迟钝 |
AR |
TECR |
|
Van Den Ende-Gupta 综合征 |
AR |
SCARF2 |
|
自身免疫性淋巴组织增生症(ALPS) |
AR |
FADD |
|
小脑共济失调 |
AD |
TGM6 |
|
逆向性痤疮 |
AD |
NCSTN |
四、方案设计
相比传统测序,外显子测序能够迅速的获得所有外显子区域的遗传信息,在大幅提升效率的同时显著降低了研究成 本;相比全基因组测序,外显子测序能够在缩短实验周期、减少数据分析量及实验投入的基础上有针对性的得到大部分全基因组测序所能得到的信息。基于外显子组 测序良好性价比,该方法目前在国际上已经被广泛的应用于遗传病和癌症研究中。
1. 单基因疾病研究方案
首先需要按照疾病表型对家系成员进行严格筛查,明确其患病情况并进行该疾病研究的背景调查。在找出该疾病已 经有一些研究背景和相关的致病基因报道,可通过传统PCR测序方法对已知的疾病相关变异进行验证和初筛;确认所研究的样本中未发现相关的基因变异,那么可 以挑选一个或数个相同疾病家系的核心成员成员进行外显子组测序。每个家系中的患病个体选取3-5个样本,正常个体选取1-2名作为对照进行研究。按照疾病 模型(AD,AR等)及样品的家系信息对测序得到的结果进行分析,缩小候选变异的范围,经过多种注释、筛选后过滤掉对功能无影响的变异及公共数据库中的常 见变异,再使用传统PCR测序进行样本扩大化验证及相关的功能研究,最终确定疾病相关变异。
单基因遗传病研究举例:
a. 家系图:
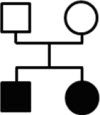
b. 分析思路:
1). 隐性纯合突变致病:两个患者共享相同的纯合突变,父母为杂合携带者。
2). 复合杂合突变致病:两个患者具有相同的突变,即在一个基因内有两个不同的杂合变异,而父母分别为这两个杂合突变的携带者。
3). 显性模式(新生突变):找两个患者共有的杂合突变,而父母不带有该突变。
c. 分析结果示意:

若样本为散发样本,由于样本间没有血缘关系,遗传背景相差较大,测序得到的结果也较难分析。为了更为准确的 得到有价值的结果,使用散发样本进行外显子组测序要求的样本数目比家系样本要多一些。一般建议至少做30个患病个体样本以上的平行测序分析。对大量患病个 体的测序数据进行多样本分析,从而确定候选疾病相关变异,再用传统PCR测序在其他的相同疾病患病个体和正常人群中做进一步验证。
2. 复杂疾病及癌症的研究方案
对于复杂疾病,首先应该选择具有遗传性较高的病例作为研究对象,一般需要满足以下几个特点:a. 与疾病相关;b. 高度遗传;c. 在患者中表现较早,表型一致,高外显率;d. 疾病的发病机制相似。整体的研究思路一般是通过适量样本的外显子测序(患病和健康个体各50例)找到与疾病高度关联的低频突变,然后根据这一结果订制合适 的芯片,在大样本里进行大规模验证。从而获得精确度更高的疾病相关变异位点。接着可以针对这些位点进行生物学功能研究,从而得到有意义的结果,开发出疾病 诊断及治疗的相关产品等。

在
各种环境因素的作用下,机体某些体细胞染色体上发生的变异破坏或改变了某些重要的生物学过程,体细胞可能会因此异常增生而转变为肿瘤细胞。由于肿瘤细胞具
有异质性,同一块肿瘤组织里可能含有不同时期的肿瘤细胞以及正常体细胞,因此它的基因变异情况相对其遗传疾病来说更为复杂。对于肿瘤组织的外显子组测序研
究,其最关键的步骤在于样本的选取。目前最常见的情况是分别取同一癌症患者的癌组织和癌旁组织进行比较,样本数目建议至少20对以上。测序后成对的样本进
行分析后再进行不同病人间的多样本分析,以此来发掘肿瘤相关的基因变异。由于肿瘤产生的原因包括基因突变,基因表达水平变异,表观遗传变异等多个方面,在
利用NGS研究肿瘤的时候,通常会使用多种试验方法相结合的方法,例如转录组测序、全基因组测序、甲基化测序等,相互进行印证,多数据整合分析可以进一步
的提高数据的可靠性,提升科研文章档次。
{nextpage}
五、捕获平台
目前主流的捕获平台,各平台的特点如下。
|
捕获平台 |
Illumina TruSeq Exome Enrichment Kit |
Roche SeqCap EZ Human Exome Library |
Agilent SureSelect Human All Exon |
|
捕获量 |
62M |
64M |
51M |
|
捕获区域 |
外显子及旁翼区, 部分UTR及miRNA |
外显子区及miRNA |
外显子区 |
|
探针 |
95 mer DNA |
90-105 mer DNA |
120 mer RNA |
|
探针数量 |
340,427 |
2,100,000 |
655,872 |
|
对常用数据库的覆盖率 |
97.2% CCDS 96.4% RefSeq 93.2% Gencode 77.6% miRBase |
99.8% CCDS 98.4% RefSeq 96.7% Gencode 98.67% miRBase |
1.22% of human genomic regions, > 700 human miRNAs, > 300 additional human non-coding RNAs |
六、项目流程

1、 样本检测
用于建库的DNA样品标准为样品浓度大于60ng/μl,体 积大于20μl,OD260/OD280为1.7-2.0。通过以下三种方式进行样本检测:
• 采用荧光定量的方法对DNA样品进行定量;
• NanoDrop检测OD260/OD280;
• 凝胶电泳检测DNA的状态,是包含蛋白质、RNA污染及是否存在DNA降解。
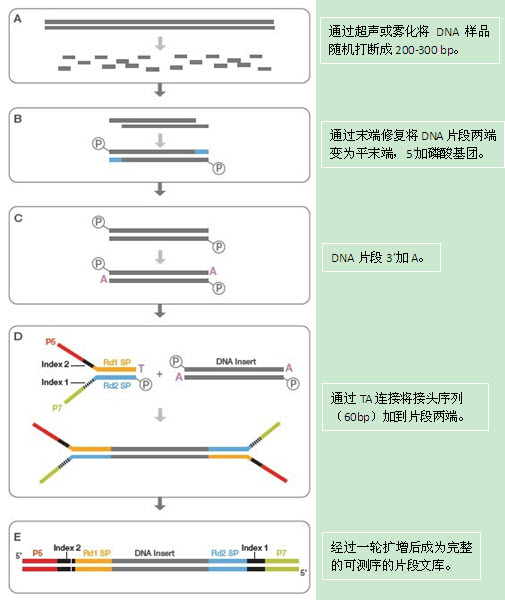
2、 建库
应用TruSeq DNA Sample Prep Kits进行文库制备,起始DNA量为1.2 μg。
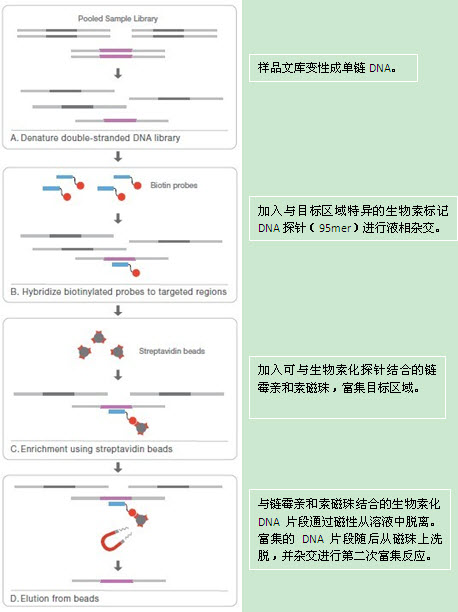
3、 捕获
以Illumina的捕获平台为例,应用TruSeq Exome Enrichment Kit捕获外显子组及旁翼区,部分UTR及miRNA,总捕获范围为62M。
4、 测序
捕获得到的DNA序列可于Illumina的任一测序仪中进行测序,以HiSeq 2000为例,每run可运行两张flowcell,每个flowcell包括8个lane,100PE模式下每run运行约11天,数据产出为 600G。捕获样品经桥式PCR后,置于flowcell中进行测序,外显子组样本一般建议测序125X,便足够进行遗传疾病分析,如肿瘤样本可根据情况 适度增加测序深度。
{nextpage}
5、 质控
严格使用Illumina原厂试剂,遵循Illumina Genome Network管理,是Illumina全球最高测序质量的代表。平均大于99% 碱基准确度达Q20,保证大于85% 碱基准确度达Q30,平均clean data占raw data 90% 以上。对于外显子组项目,约90%的外显子区域覆盖度达到10× 以上,保证最高的测序均一性。
a. 原始数据
HiSeq 2000平台产出的原始数据为Fastq格式,以下是对该格式的详细说明:
@HWI-ST1203:231:C1NDLACXX:7:1101:1837:2139 1:N:0:AGTCAA
TTCCACTTAAAAATACAAGAGCACAAATCCACATTTATTTATTGATTTTTCGTTAGTTTAAATCCTTGAGGGGTACAGCATCACTCGGATTCTGTGTCCAA
+
CCCFDFFFHHHHHJJJJIJJJJJJJJIJIJIJFHJJGJEIEIGIIJIJIIGIDGGIIHI@HHEHIIIIIJ=CHABBDFFFFEEDEEDBBDDCDDCCDDCDC
对于以上Fastq序列,第一行以@开头,后面是read的ID以及其他信息;第二行代表read的序列; 第三行一般以“+”表示;第四行代表read的质量信息,与第二行的碱基序列相对应。其中,为了便于计算机进行存储,质量值以字符来表示,每个字符所代表 的ASCII码减去33即为该碱基对应的质量值。根据相应的公式(Q=-10lgP),即可计算每个碱基被测错的概率,其中Q20代表碱基被测错的概率为 1%,Q30代表碱基被测错的概率为1‰。
将以上Fastq序列的质量信息转换成相应的质量值,结果如下。在该read中,只有一个碱基的质量值为28,其余碱基的质量值均大于30。
34,34,34,37,35,37,37,37,39,39,39,39,39,41,41,41,41,40,41,41,41,41,41,41,41,41,40,41,40,41,40,41,37,39,41,41,38,41,36,40,36,40,38,40,40,41,40,41,40,40,38,40,35,38,38,40,40,39,40,31,39,39,36,39,40,40,40,40,40,41,28,34,39,32,33,33,35,37,37,37,37,36,36,35,36,36,35,33,33,35,35,34,35,35,34,34,35,35,34,35,34.
b. 质量评估
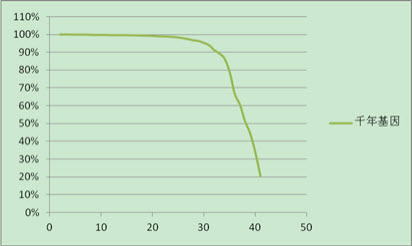
碱基质量评分
上图是基因测序得到read的质量值结果,其中几乎所有碱基的质量值在20以上,90%以上碱基的质量值在30以上。
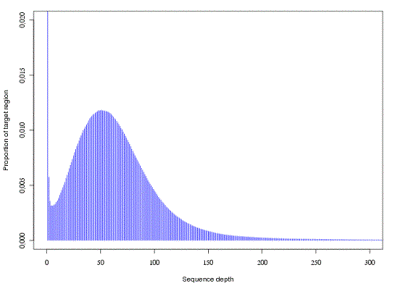
测序深度分布
虽然外显子组测序的 整体深度一般都大于100X,但由于测序过程中存在一定的序列偏向性,部分外显子区域的测序覆盖度较低。在进行信息分析时,通常只考虑测序深度高于10X 的外显子区域,以便提高分析结果的可靠性。测序结果中,85%-95%的外显子区域测序深度大于10 X,保证较高的测序均一性。
c. 结果展示
|
英文参数 |
统计结果 |
中文说明 |
|
Sample Name |
Example |
样本名 |
|
Total reads |
100,256,834 |
Reads数目 |
|
Total yield (bp) |
10,125,940,234 |
数据量 |
|
Read length (bp) |
101.0 |
读长 |
|
Target regions (bp) |
62,085,286 |
目标区域大小 |
|
Average throughput depth of target regions |
163.1 |
平均测序深度 |
|
Initial mappable reads (mapped to human genome) |
100,097,762 |
可比对序列数 |
|
% Initial mappable reads (out of total reads) |
99.8% |
可比对序列比例 |
|
Non-redundant reads (de-duplicated by Picard tools) |
82,401,028 |
非冗余序列数 |
|
% Non-redundant reads (out of initial mappable reads) |
82.3% |
非冗余序列比例 |
|
Non-redundant unique reads (uniquely mapped to human genome) |
73,028,083 |
非冗余单一比对序列数 |
|
% Non-redundant unique reads (out of non-redundant reads) |
88.6% |
非冗余单一比对序列比例 |
|
On-target reads (mapped to target regions) |
50,349,303 |
目标区域序列数 |
|
% On-target reads (out of non-redundant unique reads) |
68.9% |
目标区域序列比例 |
|
% Coverage of target regions (more than 1X) |
95.1% |
测序深度大于1×的覆盖度 |
|
Number of on-target genotypes (more than 1X) |
59,032,909 |
测序深度大于1×的区域 |
|
% Coverage of target regions (more than 10X) |
91.6% |
测序深度大于10×的覆盖度 |
|
Number of on-target genotypes (more than 10X) |
56,865,579 |
测序深度大于10×的区域 |
|
Mean read depth of target regions |
65.4 |
目标区域平均测序深度 |
|
Number of SNPs |
78,241 |
SNP数目 |
|
Number of coding SNPs |
20,593 |
编码区SNP数目 |
|
Number of synonymous SNPs |
10,654 |
同义SNP数目 |
|
Number of nonsynonymous SNPs |
9,391 |
非同义SNP数目 |
|
Number of Indels |
8,447 |
InDel数目 |
|
Number of coding Indels |
411 |
编码区InDel数目 |
6、 信息分析
a. 确定序列,原始数据过滤及统计:
通过FastQC, FastX-toolkit等软件对测序质量进行评估,去除低质量reads(大于5个碱基质量低于Q20),剩余的数据作为clean data进行分析,平均大于99%的碱基质量高于Q20,大于85%的碱基质量高于Q30。
b. Mapping:
通过bwa软件将reads map到标准参考基因组上(UCSC hg19),去除无法map到参考基因组和多重map的reads后进行后续分析,大约有99.5%的reads能进行下一轮分析。
c. 去除完全一致reads(duplicate reads):
外
显子捕获过程中含有PCR扩增步骤,会人为引入完全一致的DNA片段,由于这些DNA序列会对后期的分析造成影响,故要使用PICARD软件去除数据中的
duplicate reads,不同的捕获平台中这类序列所占的比例不一样,illumina捕获平台中的duplicate
reads数目约占总数据的15-20%,Agilent平台中的这一数值约为1-3%。
d. 对目标区域内的序列进行变异检出:
使用Samtools对测序结果与参考基因组进行比对,找出样品中存在的变异,包括SNV,InDel等,并对其进行注释及功能预测,包括dbSNP、1000G数据库,SIFT,Polyphen-2,GERP等软件。
e. 多样本分析:
根据研究内容的不同,将多个样本分为不同的组别,对其中的变异信息进行汇总,统计变异在群体内出现的频率,位置等相关信息,通过KEGG等信号通路注释分析其与疾病之间的关联。
f. 报告提交:
包括样品检测与建库报告(pdf格式)、测序结果报告(pdf格式)、单样本变异检出报告(excel格式)、多样本汇总分析报告(excel格式)、原始数据(fastq、BAM等格式)和发表文章所需的各类图表。
七、外显子组测序相关名词
外显子组测序:是指利用序列捕获技术将全基因组外显子区域DNA捕捉并富集后进行高通量测序的基因组分析方法。外显子测序相对于基因组重测序成本较低,对研究已知基因的SNP、 InDel 等具有较大的优势。
测序深度:测序得到的总碱基数与待测区域大小的比值。如使用Illumina TruSeq Exome Enrichment Kit,该试剂盒的捕获范围为62M,测序得到620M数据量时,测序深度为620/62=10×。
覆盖度:指测序获得的序列占整个待测区域的比例。如果外显子组测序的覆盖度是98%,则表示仍有2%的序列区域是没有通过测序获得的。
Read:就是读长,就是高通量测序时一个反应所能测出的碱基数。
SNP(single nucleotide polymorphism):单核苷酸多态性,个体间基因组DNA序列同一位置单个核苷酸变异(替代、插入或缺失)所引起的多态性;不同物种个体基因组 DNA 序列同一位置上的单个核苷酸存在差别的现象。
InDel(Insertion/Deletion):插入/缺失,是指两种亲本在全基因组中的差异,相对另一个亲本而言,其中一个亲本的基因组中有一定数量的核苷酸插入或缺失。
CNV(copy number variation):基因组拷贝数变异,是基因组变异的一种形式,通常使基因组中大片段的DNA形成非正常的拷贝数量。
SV(structure
variation):基因组结构变异,染色体结构变异是指在染色体上发生了大片段的变异。主要包括染色体大片段的插入和缺失(引起 CNV
的变化),染色体内部的某块区域发生重复复制、翻转颠换、易位、两条染色体之间发生重组(inter-chromosome
trans-location)等。
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言




#外显子组测序#
48
#外显子组#
40
#外显子#
44