好文推荐 | 小胶质细胞髓样细胞触发受体2及其可溶形式在肌萎缩侧索硬化症中的研究进展
2023-11-01 中风与神经疾病杂志 中风与神经疾病杂志 发表于上海

小胶质细胞作为CNS内主要的免疫细胞,在肌萎缩侧索硬化症疾病进展过程中发挥着重要的作用,现将小胶质细胞TREM2、sTREM2及其在ALS中的研究进行综述。
髓样细胞触发受体2(triggering receptor expressed on myeloid cells 2,TREM2)是一种表达在髓系细胞上的跨膜受体,在中枢神经系统(central nervous system,CNS)内,仅表达在小胶质细胞表面,参与小胶质细胞的一系列活动,包括扩增、迁移、存活、激活、吞噬,具有抑制炎症反应、促进小胶质细胞吞噬病理性蛋白、凋亡神经元等作用。可溶形式TREM2(soluble TREM2,sTREM2)是TREM2经蛋白水解或异常的转录本翻译而来,可在血液及脑脊液中检测,不仅预示着TREM2的存在,也存在生物学作用,参与小胶质细胞的功能活动。小胶质细胞作为CNS内主要的免疫细胞,在肌萎缩侧索硬化症(amyotrophic lateral sclerosis,ALS)疾病进展过程中发挥着重要的作用,现将小胶质细胞TREM2、sTREM2及其在ALS中的研究进行综述。
1 TREM2及sTREM2
1.1 TREM2基因结构 TREM2由位于人类染色体6p21.1上的TREM2基因所编码。TREM2蛋白由胞外结构域(由外显子1-3编码,其中外显子1编码一种信号肽,外显子2编码类免疫球蛋白结构域)、跨膜结构域(由外显子4编码)和胞内结构域(由外显子5编码)组成,细胞信号传导由TREM2蛋白的三个区域共同调节。在人脑中已经报道了四种主要的TREM2基因转录本:即ENST00000373113、ENST00000373122、ENST00000338469和TREM2Δe2。ENST00000373113是表达量最多、最长的TREM2转录本,包含上述所有5个外显子,编码全长230个氨基酸的跨膜受体蛋白。ENST00000373122没有外显子5,是第二长的转录本(编码222个氨基酸),并且其外显子4包含一个能改变其编码序列的替代起点。ENST00000338469(编码219个氨基酸)缺失了编码跨膜区的外显子4。TREM2Δe2则缺少外显子2(编码类免疫球蛋白结构域),但保留了其他外显子,最终在膜上产生一个没有与配体结合能力的非功能性受体。但值得注意的是目前尚不清楚ENST00000373122、ENST00000338469和TREM2Δe2这三种转录本是否在机体内被翻译。
1.2 TREM2的配体及其下游信号通路 TREM2是免疫球蛋白超家族的跨膜受体,可以与多种配体相互作用,包括细菌产物、DNA、脂蛋白和磷脂等。一些配体在生理条件下存在于体内,例如低密度脂蛋白(low-density lipoprotein,LDL)和载脂蛋白(apolipoproteins,APOE)。在组织损伤和细胞死亡的过程中,也会释放相应的TREM2配体,例如,在细菌感染的情况下,TREM2通过各种表面磷脂(磷脂酰丝氨酸、心磷脂等)和糖脂(硫脂、其他脑苷脂等)结合细菌阴离子分子,如脂多糖(lipopolysaccharide,LPS)、葡聚糖硫酸盐以及细胞碎片;在阿尔茨海默病(Alzheimer’s disease,AD)的大脑中,TREM2可以直接与病理性β-淀粉样蛋白(β-amyloid,Aβ)寡聚体相互作用。TREM2因缺乏免疫受体酪氨酸的活化基序(immunoreceptor tyrosine-based activation motif,ITAM)需要与相应的共受体结合来发挥作用。对小鼠巨噬细胞的研究表明,TREM2与接头蛋白DNAX激活蛋白12(DNAX activation protein 12,DAP12)和DAP10(DNAX activation protein 10,DAP10)通过跨膜区的相反电荷残基结合。当TREM2与配体相互作用时,这些共受体被磷酸化,形成SH2的结合位点,并招募细胞内的信号传导机制。DAP12,也被称为酪氨酸激酶结合蛋白(TYRO protein tyrosine kinase binding protein,TYROBP),介导脾酪氨酸激酶Syk的激活,而DAP10通过招募磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)来促进信号的传导。TREM2可与DAP12或DAP10结合,并形成TREM2-DAP12/DAP10异二聚体。在小鼠巨噬细胞中,DAP12是钙动员所必需的,而DAP10是激活丝氨酸/苏氨酸蛋白激酶和细胞外信号调节激酶(extracellular signal-regulated kinase,ERK)的关键。除上述传导过程外,其他信号通路也可能调节TREM2的信号传导。首先,DAP12的ITAM基序可以在集落刺激因子1受体(colony stimulating factor 1 receptor,CSF1R)激活时被SRC酪氨酸激酶磷酸化,因此,该信号的活性也将调节TREM2的信号传导。其次,参与吞噬凋亡碎片的TAM受体(TYRO3,AXL,MER)也可以参与表达TREM2的小胶质细胞的活动,但他们之间的相互作用尚不清楚。此外,小胶质细胞主要存在两种不同的吞噬受体类型,一种是对外来微生物病原体具有高亲和力的Toll样受体(Toll-like receptor,TLR),另一种是可以识别凋亡的细胞物质的受体TREM2。有研究表明,通过DAP12的TREM2信号传导可拮抗TLR的表达以及TLR介导的炎症细胞因子的产生;相反,TREM2的表达可被LPS(作为TLR4的配体)或干扰素γ(interferon gamma,IFNγ)的促炎信号所抑制。最后,有研究表明刺激核受体过氧化物酶体增殖物激活受体(nuclear receptors peroxisome proliferator activated receptor,PPARγ and PPARδ)、肝X受体(liver X receptor,LXR)和维甲酸X受体(retinoid X receptor,RXR)可以促进小鼠TREM2和其他吞噬相关受体的表达。总之,TREM2信号通路是一个复杂的过程,TREM2信号在体内调控需要进一步的研究。
1.3 sTREM2的产生 2008年,Piccio等首次在人类脑脊液和血清中发现了sTREM2的存在。目前关于sTREM2如何产生,推测存在以下两种独立或协作的过程:TREM2胞外区经蛋白水解切割和脱落以及缺乏跨膜区的选择性剪接的TREM2转录本的翻译。去整合素和金属蛋白酶 (a disintegrin and metalloprotease,ADAM)家族的成员,主要是ADAM10和ADAM17,在组氨酸157和丝氨酸158(H157-S158)之间催化TREM2胞外结构域的脱落。ADAM17在THP1和CHO细胞系中sTREM2的产生起主要作用,而ADAM10与人类巨噬细胞、HEK293细胞和小鼠小胶质细胞中sTREM2的脱落有关。也有研究发现在巨噬细胞中蛋白酶meprin β可以在精氨酸136和天冬氨酸137之间(R136-D137)裂解TREM2,从而释放sTREM2。TREM2转录本ENST00000338469不包括编码跨膜区的外显子4,约占大脑中总TREM2 mRNA的25%,因此,大约20%~25%的总sTREM2蛋白可能来自该转录本的翻译,而不是通过脱落酶活性切割全长的跨膜TREM2产生。
1.4 sTREM2水平的调控及其影响因素 生理条件下,全长TREM2在巨噬细胞上的周转非常迅速,半衰期不到1 h,快速、可诱导的产生sTREM2。sTREM2的脱落通常发生在TREM2与配体结合后,并且新合成的TREM2蛋白需要不断成熟并运输到细胞表面,以维持持续的受体活性。因此,sTREM2的水平可能反映了TREM2受体与其配体的结合程度、其脱落以及新的TREM2蛋白生成及运输的速度。关于增加或减少sTREM2水平的因素目前尚不清楚。有研究表明,LPS或白介素1β可以诱导小鼠原代小胶质细胞释放sTREM2;其他细胞因子如白介素13或白介素4也可以诱导sTREM2脱落;此外,Aβ寡聚体也可以诱导过表达TREM2的细胞释放sTREM2。sTREM2水平受到基因及人口学因素的影响:关于基因方面,TREM2受体类免疫球蛋白结构域内的突变,如p. T66M和p. Y38C会导致TREM2的错误折叠,导致未成熟蛋白质保留在内质网中,进而使得细胞表面TREM2水平降低,脱落减少,因而体外检测到sTREM2水平降低,而在携带p. R47H变异者中检测到的脑脊液sTREM2水平却显著高于非携带者;p. H157Y变异增加了TREM2的脱落,同样也会导致sTREM2水平升高;此外,有研究表明,11号染色体MS4A基因座附近的变异与脑脊液sTREM2水平具有相关性。在人口学方面,包括年龄、性别和种族,也可能会影响脑脊液和血液sTREM2水平,因而在评估sTREM2在不同神经系统疾病中的作用时,应该在分析和研究设计中加以考虑。
2 TREM2、sTREM2在CNS中的功能及其在ALS中的研究
在CNS内,TREM2仅表达在小胶质细胞上,参与小胶质细胞的扩增、迁移、存活、激活、吞噬,维持小胶质细胞的能量代谢及脂代谢,抑制炎症反应。TREM2基因突变与许多神经系统退行性疾病有关,包括ALS。对ALS小鼠模型和ALS患者死后组织的空间转录组学分析显示小胶质细胞功能障碍早在ALS症状出现之前就已发生,且TREM2信号是小胶质细胞激活的早期步骤,TREM2在反应性小胶质细胞中被上调。而sTREM2不仅预示着TREM2的存在,也存在一定的生物学作用,参与小胶质细胞的功能活动。
2.1 TREM2
2.1.1 调节小胶质细胞吞噬功能 TREM2是小胶质细胞吞噬活动的重要调节因子。在AD中,小胶质细胞直接接触和吞噬Aβ需要TREM2的介导。在脱髓鞘模型中,TREM2激动剂AL002a可以促进小胶质细胞对髓鞘的摄取和处理。TREM2还可以与神经元上的内源性配体相互作用,并介导对凋亡神经元细胞的吞噬。小鼠和体外培养中TREM2的缺失或损伤减少了小胶质细胞对凋亡细胞、细胞碎片、脂蛋白和细菌的吞噬。在非吞噬细胞中的过表达TREM2,如CHO细胞,可诱导其对细菌、脂蛋白和细胞碎片的吞噬。值得注意的是,有研究发现用ADAM抑制剂阻止TREM2的脱落可以促进小胶质细胞对髓鞘和Aβ的摄取,因而依赖TREM2的吞噬活动似乎需要全长的TREM2的作用。在Xie等的研究中,利用ALS小鼠TDP-43蛋白病变模型,发现TREM2缺陷的小胶质细胞失去了对TDP-43包涵体的吞噬能力,表明TREM2在TDP-43诱导的神经变性中具有保护作用。TREM2介导的底物识别和吞噬的确切机制尚不清楚,推测TREM2可能作为吞噬底物的受体,直接与它们结合。
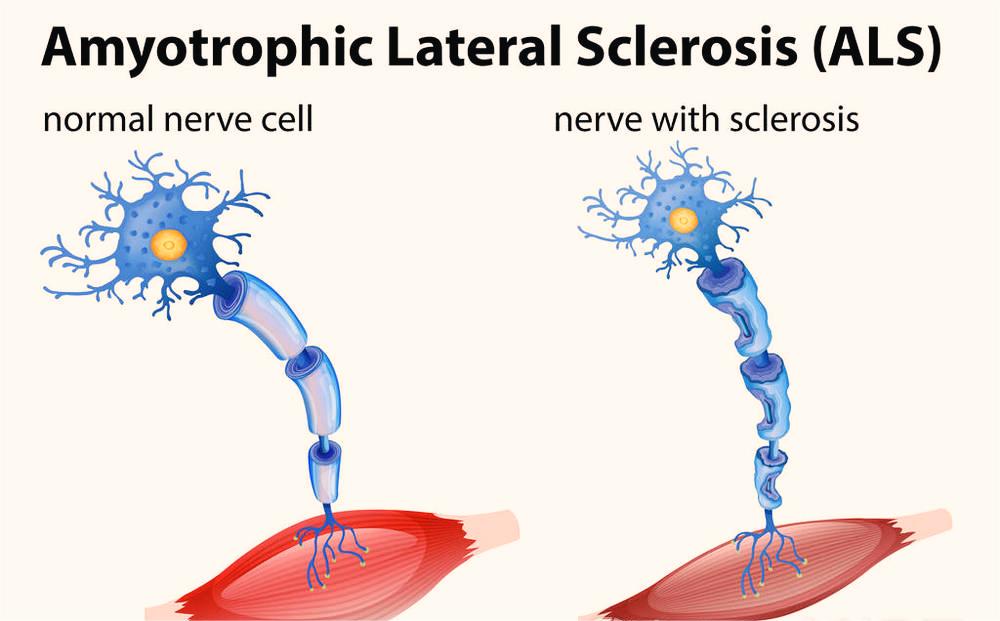
2.1.2 参与小胶质细胞代谢 小胶质细胞依赖糖酵解和氧化代谢来提供能量并维持其功能,TREM2通过哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)信号通路维持小胶质细胞能量和生物合成代谢,这可能是TREM2缺陷的小胶质细胞对错误折叠的蛋白质和损伤的细胞器吞噬能力降低的原因。此外,TREM2也被证明可以调节小胶质细胞的脂质代谢,在脱髓鞘模型中,TREM2缺陷的小胶质细胞虽然可以正常吞噬髓鞘,但却无法消化髓鞘脂质,进一步应用单细胞RNA测序分析发现在TREM2缺陷的小胶质细胞中,存在溶酶体降解和胆固醇运输相关的转录异常。Kang等在SOD1(G93A)ALS小鼠模型中观察到轴突进行性脱髓鞘,并且早于疾病发生。TREM2参与脂质代谢是否与ALS的慢性脱髓鞘有关需要进一步的研究来证实。
2.1.3 抑制炎症反应 TREM2的激活可以拮抗髓系细胞对促炎刺激的反应。体外和体内研究均表明TREM2具有抗炎作用。体外敲除TREM2削弱了白介素4诱导的原代小胶质细胞的抗炎反应,并在LPS治疗后增强了促炎介质的表达,包括诱导型一氧化氮合酶、肿瘤坏死因子α、白介素1β和白介素6;体内研究表明,TREM2通过抑制炎症反应,进而减轻神经炎症诱导的P301S小鼠内过度激活的tau激酶,而减少TREM2的表达加剧了SAMP8小鼠的衰老相关的神经炎症。其次,在分子水平上,小鼠的小胶质细胞和巨噬细胞中几个标志性的抗炎基因以TREM2依赖的方式表达,包括Galectin-1和Galectin-3、白细胞介素1受体拮抗剂、原颗粒蛋白等。此外,上文中提到通过DAP12的TREM2信号可拮抗TLR的表达以及TLR介导的炎症细胞因子的产生,进而具有抑制炎症的作用。CNS炎症是ALS的特征之一,对41个来自散发性ALS患者的运动皮质样本的全基因组特征分析显示:总共有1573个与炎症相关的基因发生了变化。此外,也有研究表明血液和脑脊液中的炎症指标与ALS存活率具有相关性。因此,靶向TREM2以调节炎症可能是治疗ALS的有效方法。
2.2 sTREM2s TREM2的脱落对TREM2受体信号的影响,以及sTREM2本身的内源性功能,目前尚不完全清楚,sTREM2在细胞外释放后,可通过以下方式发挥作用:(1)由于胞外结构域脱落和缺乏与配体结合能力而抑制TREM2受体的信号通路;(2)与其他细胞(即星形胶质细胞、神经元、巨噬细胞)或小胶质细胞本身的未知受体结合,从而以自分泌方式发挥作用;(3) 阻止特定配体与膜TREM2受体的进一步结合,最终抑制TREM2信号传导。越来越多的研究表明脑脊液中sTREM2水平不但是小胶质细胞激活的生物标志物,并且其本身即可以导致小胶质细胞激活。sTREM2在体内和体外均具有功能效应:直接或通过腺病毒介导将sTREM2注入5XFAD转基因小鼠(一种Aβ积聚的AD小鼠模型)的海马区,sTREM2可以增强小胶质细胞的增殖、迁移和聚集在斑块周围,与Aβ结合并且可以抑制Aβ聚集,降低Aβ负荷;在原代培养的小鼠小胶质细胞中,sTREM2可以不依赖全长TREM2,抑制细胞自噬,从而促进小胶质细胞存活并刺激依赖于核因子-κB的炎性细胞因子的产生;将sTREM2立体定向注射到野生型或TREM2基因敲除小鼠的海马区后,诱导了小胶质细胞激活的形态变化。脑脊液中sTREM2的浓度与AD患者脑脊液中总tau蛋白和磷酸化tau蛋白水平具有相关性,提示脑脊液sTREM2水平可以作为神经元损伤的替代免疫生物标志物。
在ALS患者中,脑脊液sTREM2水平显著升高。这种升高在ALS的早期阶段最为明显。在疾病晚期,sTREM2水平与病程呈正相关,较高水平的sTREM2与疾病晚期进展缓慢相关,表明sTREM2具有潜在的保护作用。
3 治疗启示及未来的研究方向
3.1 治疗启示 TREM2基因突变可能会增加患ALS的风险,因此,纠正突变的TREM2基因可能是预防ALS的一种合理的治疗策略。除了以基因组为靶点外,应用外来小胶质细胞样细胞取代小鼠体内的小胶质细胞,或者用TREM2过表达的诱导性多能干细胞来源的小胶质细胞替代内源性小胶质细胞,可能会降低TREM2变异携带者发生ALS的风险或推迟疾病的发病时间。除此之外,应用药物干预或基因治疗增加小胶质细胞TREM2的活性或表达也可能会缓解ALS相关的功能障碍,如TREM2的激动型单抗或反义寡核苷酸(Antisense oligonucleotide,ASO)。有研究发现将纯化的sTREM2直接注射或应用表达sTREM2的病毒载体均对AD小鼠模型具有保护作用,因此,此方法也可能对患有ALS的患者具有一定的保护作用。
3.2 未来的研究方向 疾病相关小胶质细胞(disease-associated microglia,DAM)的特点是稳态基因下调,参与吞噬、脂质代谢和溶酶体途径的相关基因上调,且DAM具有神经保护作用,可能作为神经变性相关分子模式的传感器,识别包括死亡神经元、髓鞘碎片和蛋白质聚集体上的特定分子,进而抑制神经退行性病变。转录研究显示,ALS中出现了DAM,且与TREM2的表达呈正相关,然而,目前对DAM在ALS中所起的作用了解有限,需要进一步的研究来确定诱发DAM的具体诱因、TREM2信号如何在ALS中诱导DAM,以及在不同的疾病阶段如何改变,TREM2介导的DAM在所有类型的ALS中是否存在。
目前关于sTREM2的确切功能以及其是否参与中枢神经系统病理依然存在争议:sTREM2通过改善小胶质细胞的清除作用而发挥神经保护作用,但sTREM2也可以触发小胶质细胞释放促炎细胞因子,这可能会对神经元功能产生不利影响;用ADAM抑制剂阻止TREM2的脱落,可以促进小胶质细胞的吞噬活动,sTREM2直接注射或应用表达sTREM2的病毒载体对AD小鼠模型却具有保护作用。此外,关于sTREM2的配体和受体的了解也有限,因此,未来需要进行更为系统的研究。
4 总结
TREM2及sTREM2参与小胶质细胞的功能活动,与ALS疾病过程有关,靶向小胶质细胞TREM2及sTREM2可能是治疗ALS的一种的途径,但我们对其在ALS不同疾病阶段或不同ALS病理中的作用的了解仍然有限,因此未来需要更多深入的研究。
参考文献
[1]Filipello F,Goldsbury C,You SF,et al. Soluble TREM2:innocent bystander or active player in neurological diseases[J]. Neurobiol Dis,2022,165(4):105630.
[2]Takahashi K,Rochford CDP,Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2[J]. J Exp Med,2005,201(4):647-657.
[3]Zhong L,Chen XF,Zhang ZL,et al. DAP12 stabilizes the c-terminal fragment of the triggering receptor expressed on myeloid cells 2 (TREM2) and protects against LPS-induced pro-inflammatory response[J]. J Biol Chem,2015,290(25):15866-15877.
[4]Mazaheri F,Snaidero N,Kleinberger G,et al. TREM2 deficiency impairs chemotaxis and microglial responses to neuronal injury[J]. EMBO Rep,2017,18(7):1186-1198.
[5]Wang Y,Cella M,Mallinson K,et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model[J]. Cell,2015,160(6):1061-1071.
[6]Qin Q,Teng Z,Liu C,et al. TREM2,microglia,and Alzheimer’s disease[J]. Mech Ageing Dev,2021,195(4):111438.
[7]Brendel M,Kleinberger G,Probst F,et al. Increase of TREM2 during aging of an Alzheimer’s disease mouse model is paralleled by microglial activation and amyloidosis[J]. Front Aging Neurosci,2017,9(1):8.
[8]Ulland TK,Song WM,Huang SCC,et al. TREM2 maintains microglial metabolic fitness in Alzheimer’s disease[J]. Cell,2017,170(4):649-663.
[9]Deczkowska A,Weiner A,Amit I. The physiology,pathology,and potential therapeutic applications of the TREM2 signaling pathway[J]. Cell,2020,181(6):1207-1217.
[10]Liu W,Taso O,Wang R,et al. Trem2 promotes anti-inflammatory responses in microglia and is suppressed under pro-inflammatory conditions[J]. Hum Mol Genet,2020,29(19):3224-3248.
[11]Yin J,Liu X,He Q,et al. Vps35-dependent recycling of Trem2 regulates microglial function[J]. Traffic,2016,17(12):1286-1296.
[12]Jiang T,Tan L,Zhu XC,et al. Silencing of TREM2 exacerbates tau pathology,neurodegenerative changes,and spatial learning deficits in P301S tau transgenic mice[J]. Neurobiol Aging,2015,36(12):3176-3186.
[13]Hsieh CL,Koike M,Spusta SC,et al. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia[J]. J Neurochem,2009,109(4):1144-1156.
[14]Xie M,Liu YU,Zhao S,et al. TREM2 interacts with TDP-43 and mediates microglial neuroprotection against TDP-43-related neurodegeneration[J]. Nat Neurosci,2022,25(1):26-38.
[15]Piccio L,Buonsanti C,Cella M,et al. Identification of soluble TREM2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation[J]. Brain,2008,131(Pt 11):3081-3091.
[16]Zhong L,Xu Y,Zhuo R,et al. Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer’s disease model[J]. Nat Commun,2019,10(1):1365.
[17]Zhong L,Chen XF,Wang T,et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival[J]. J Exp Med,2017,214(3):597-607.
[18]Bekris LM,Khrestian M,Dyne E,et al. Soluble TREM2 and biomarkers of central and peripheral inflammation in neurodegenerative disease[J]. J Neuroimmunol,2018,319:19-27.
[19]Rauchmann BS,Sadlon A,Perneczky R,et al. Soluble TREM2 and Inflammatory Proteins in Alzheimer’s disease cerebrospinal fluid[J]. J Alzheimers Dis,2020,73(4):1615-1626.
[20]Maniatis S,ij T,Vickovic S,et al. Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis[J]. Science,2019,364(6435):89-93.
[21]Yang J,Fu Z,Zhang X,et al. TREM2 ectodomain and its soluble form in Alzheimer’s disease[J]. J Neuroinflammation,2020,17(1):204.
[22]Jin SC,Benitez BA,Karch CM,et al. Coding variants in TREM2 increase risk for Alzheimer’s disease[J]. Hum Mol Genet,2014,23(21):5838-5846.
[23]Kiianitsa K,Kurtz I,Beeman N,et al. Novel TREM2 splicing isoform that lacks the V-set immunoglobulin domain is abundant in the human brain[J]. J Leukoc Biol,2021,110(5):829-837.
[24]Del-Aguila JL,Benitez BA,Li Z,et al. TREM2 brain transcript-specific studies in AD and TREM2 mutation carriers[J]. Mol Neurodegener,2019,14(1):18.
[25]Zhong L,Wang Z,Wang D,et al. Amyloid-beta modulates microglial responses by binding to the triggering receptor expressed on myeloid cells 2 (TREM2)[J]. Mol Neurodegener,2018,13(1):15.
[26]Peng Q,Malhotra S,Torchia JA,et al. TREM2-and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1[J]. Sci Signal,2010,3(122):ra38.
[27]Otero K,Shinohara M,Zhao H,et al. TREM2 and β-catenin regulate bone homeostasis by controlling the rate of osteoclastogenesis[J]. J Immunol,2012,188(6):2612-2621.
[28]Otero K,Turnbull IR,Poliani PL,et al. Macrophage colony-stimulating factor induces the proliferation and survival of macrophages via a pathway involving DAP12 and beta-catenin[J]. Nat Immunol,2009,10(7):734-743
[29]Savage JC,Jay T,Goduni E,et al. Nuclear receptors license phagocytosis by Trem2+ myeloid cells in mouse models of Alzheimer’s disease[J]. J Neurosci,2015,35(16):6532-6543.
[30]Hamerman JA,Jarjoura JR,Humphrey MB,et al. Cutting edge:inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)-2 and DAP12[J]. J Immunol,2006,177(4):2051-2055.
[31]Gao X,Dong Y,Liu Z,et al. Silencing of triggering receptor expressed on myeloid cells-2 enhances the inflammatory responses of alveolar macrophages to lipopolysaccharide[J]. Mol Med Rep,2013,7(3):921-926
[32]Feuerbach D,Schindler P,Barske C,et al. ADAM17 is the main sheddase for the generation of human triggering receptor expressed in myeloid cells (hTREM2) ectodomain and cleaves TREM2 after Histidine 157[J]. Neurosci Lett,2017,660(11):109-114.
[33]Thornton P,Sevalle J,Deery MJ,et al. TREM2 shedding by cleavage at the H157-S158 bond is accelerated for the Alzheimer’s disease-associated H157Y variant[J]. EMBO Mol Med,2017,9(10):1366-1378.
[34]Schlepckow K,Kleinberger G,Fukumori A,et al. An Alzheimer-associated TREM2 variant occurs at the ADAM cleavage site and affects shedding and phagocytic function[J]. EMBO Mol Med,2017,9(10):1356-1365.
[35]Berner DK,Wessolowski L,Armbrust F,et al. Meprin β cleaves TREM2 and controls its phagocytic activity on macrophages[J]. FASEB J,2020,34(5):6675-6687.
[36]Wu K,Byers DE,Jin X,et al. TREM-2 promotes macrophage survival and lung disease after respiratory viral infection[J]. J Exp Med,2015,212(5):681-697.
[37]Vilalta A,Zhou Y,Sevalle J,et al. Wild-type sTREM2 blocks Aβ aggregation and neurotoxicity,but the Alzheimer’s R47H mutant increases Aβ aggregation[J]. J Biol Chem,2021,296:100631.
[38]Kleinberger G,Yamanishi Y,Suárez-Calvet M,et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis[J]. Sci Transl Med,2014,6(243):243ra86.
[39]Piccio L,Deming Y,Del-águila JL,et al. Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status[J]. Acta Neuropathol,2016,131(6):925-933.
[40]Deming Y,Filipello F,Cignarella F,et al. The MS4A gene cluster is a key modulator of soluble TREM2 and Alzheimer’s disease risk[J]. Sci Transl Med,2019,11(505):eaau2291.
[41]Nugent AA,Lin K,Lengerich VB,et al. TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge[J]. Neuron,2020,105(5):837-854.e9.
[42]Cady J,Koval ED,Benitez BA,et al. TREM2 variant p. R47H as a risk factor for sporadic amyotrophic lateral sclerosis[J]. JAMA Neurol,2014,71(4):449-453.
[43]Parhizkar S,Arzberger T,Brendel M,et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE[J]. Nat Neurosci,2019,22(2):191-204.
[44]Cignarella F,Filipello F,Bollman B,et al. TREM2 activation on microglia promotes myelin debris clearance and remyelination in a model of multiple sclerosis[J]. Acta Neuropathol,2020,140(4):513-534.
[45]Schlepckow K,Monroe KM,Kleinberger G,et al. Enhancing protective microglial activities with a dual function TREM2 antibody to the stalk region[J]. EMBO Mol Med,2020,12(4):e11227.
[46]Ulland TK,Song WM,Huang S C,et al. TREM2 maintains microglial metabolic fitness in Alzheimer’s disease[J]. Cell,2017,170(4):649-663.e13.
[47]Kang SH,Li Y,Fukaya M,et al. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis[J]. Nat Neurosci,2013,16(5):571-579.
[48]Giovanna M,Gianmaria SA,Sebastiano C. Neuroinflammation and ALS:transcriptomic insights into molecular disease mechanisms and therapeutic targets[J]. Mediators Inflamm,2017,2017:1-9.
[49]Olesen M N,Wuolikainen A,Nilsson AC,et al. Inflammatory profiles relate to survival in subtypes of amyotrophic lateral sclerosis[J]. Neurol Neuroimmunol Neuroinflamm,2020,7(3):e697.
[50]Cooper-Knock J,Green C,Altschuler G,et al. A data-driven approach links microglia to pathology and prognosis in amyotrophic lateral sclerosis[J]. Acta Neuropathol Commun,2017,5(1):23.
[51]Xu Z,Rao Y,Huang Y,et al. Efficient strategies for microglia replacement in the central nervous system[J]. Cell Rep,2020,33(8):108443.
[52]Abud EM,Ramirez RN,Martinez ES,et al. IPSC-derived human microglia-like cells to study neurological diseases[J]. Neuron,2017,94(2):278-293,e9.
[53]Schoch KM,Ezerskiy LA,Morhaus MM,et al. Acute Trem2 reduction triggers increased microglial phagocytosis,slowing amyloid deposition in mice[J]. Proc Natl Acad Sci U S A,2021,118(27):e2100356118.
[54]Wang S,Mustafa M,Yuede CM,et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model[J]. J Exp Med,2020,217(9):e20200785.
[55]Keren-Shaul H,Spinrad A,Weiner A,et al. A unique microglia type associated with restricting development of Alzheimer’s disease[J]. Cell,2017,169(7):1276-1290.e17.
[56]Deczkowska A,Keren-Shaul H,Weiner A,et al. Disease-associated microglia:a universal immune sensor of neurodegeneration[J]. Cell,2018,173(5):1073-1081.
[57]Dols-Icardo O,Montal V,Sirisi S,et al. Motor cortex transcriptome reveals microglial key events in amyotrophic lateral sclerosis[J]. Neurol Neuroimmunol Neuroinflamm,2020,7(5):e829.
[58]Tam OH,Rozhkov NV,Shaw R,et al. Postmortem cortex samples identify distinct molecular subtypes of ALS:retrotransposon activation,oxidative stress,and activated glia[J]. Cell Rep,2019,29(5):1164-1177.e5.
作者信息
基金项目: 河北省自然科学基金资助项目(编号:H2021206310)
作者单位: (河北医科大学第二医院神经内科,河北 石家庄 050000)
通讯作者: 刘亚玲,E-mail:lyldoctor@163.com
引证本文
田梅,刘亚玲. 小胶质细胞髓样细胞触发受体2及其可溶形式在肌萎缩侧索硬化症中的研究进展[J]. 中风与神经疾病杂志,2023,40(1):6-10.

本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言